- Published:
- 20 January 2025
- Author:
- Matthew Clarke and Tom Jacques
- Read time:
- 15 Mins
Brain tumours in children have a devastating impact through the years of life lost and life-altering disability. Pathology is the principal step in determining their outcomes because it is the major determinant of treatments that define the likelihood of survival and damage to the developing brain.
Paediatric brain tumours differ from adult brain tumours in that they are morphologically and genetically distinct and driven by different gene pathways and mutations. Moreover, their clinical management differs due to the vulnerability of the developing brain.
The field has changed dramatically in recent years, principally due to the routine use of methylation profiling, which has redefined many tumour types, discovered many more and reframed diagnostic approaches for all. This occurs against the challenges of delivering equity of access (where experience is unevenly distributed), in a specialty not recognised in many countries, and using technology that requires capital investment unaffordable in much of the world. Paediatric neuropathology can be seen as a prototype for the challenges and opportunities of specialist pathology.
Brain tumours are the most common cause of death in children and young people after infancy. Those who survive risk lifelong impairments. Cancer Research UK data states that 76.6% of children (up to 14 years) with a central nervous system (CNS) tumour survive for 5 years or more (2012–2016). Among the survivors, 62% have at least 1 impairment, including motor and visual impairments and epilepsy.1 There are also cumulative risks of cognitive and neurological impairment that children face as they get older.2
The reasons for these poor outcomes are complex, but a common theme is the challenges and impacts of treating the developing brain with aggressive therapies, such as adjuvant craniospinal radiotherapy. Long-term treatment-related disability is in the balance with survival when stratifying treatment.
Pathology has a critical role; there are many types and subtypes of paediatric CNS tumours that require different treatment protocols that balance that risk. When determining the type and subtype of a child’s brain tumour, the pathologist is walking a tightrope between over-treatment (and, therefore, disability), which, in contrast to many more common tumours, may have an impact for 5, 6, 7 or more decades, and under-treatment (and mortality).
Since its 2007 edition, the WHO classification has evolved to include new paediatric-specific tumour types and subtypes (Table 1).3–5 The main drivers of this expansion are the discovery and refinement of tumour types by molecular characterisation, particularly the frequent use of DNA methylation profiling. The speed at which new tumours are discovered has proven a major challenge to clinical practice and has led to the establishment of the c-IMPACT-NOW Consortium, which publishes interim guidance between editions of the WHO.
Table 1: The number of paediatric-type brain tumour entities occurring in the different iterations of the WHO Classification of CNS tumours.
| WHO CNS classification | Number of paediatric brain tumour entities |
| 2007 | 23 |
| 2016 | 35 |
| 2021 | 43 |
We are experiencing a change in our diagnostic approach, driven by a shift towards molecular characterisation, with many of the tumours featured in the latest iteration of the WHO classification defined by their molecular features.
This has also led the community to reflect on what a diagnosis is – for example, which test defines the ‘ground truth’ in the diagnostic pathway. Change can be challenging to implement; the neuro-oncology community frequently encounters unique challenges in how to treat and prognosticate these tumours. Also, the accessibility of the required diagnostic tests is vital; all paediatric brain tumours should undergo the necessary molecular workup. However, inequalities in access can mean the standard can be difficult to meet.
Table 2: The different paediatric CNS tumour entities, as per the 5th edition of the WHO classification of paediatric tumours.6
| Gliomas, glioneuronal tumours and neuronal tumours |
Paediatric-type diffuse low-grade gliomas Diffuse astrocytoma, MYB- or MYBL1-altered Paediatric-type diffuse high-grade gliomas Diffuse midline glioma, H3 K27-altered Circumscribed astrocytic gliomas Pilocytic astrocytoma Glioneuronal and neuronal tumours Ganglioglioma |
| Ependymal tumours | Supratentorial ependymoma Supratentorial ependymoma, ZFTA fusion-positive Supratentorial ependymoma, YAP1 fusion-positive Posterior fossa ependymoma Posterior fossa group A (PFA) ependymoma Posterior fossa group B (PFB) ependymoma Spinal ependymoma Spinal ependymoma, MYCN-amplified Myxopapillary ependymoma |
| Choroid plexus tumours | Choroid plexus papilloma Atypical choroid plexus papilloma Choroid plexus carcinoma |
| CNS embryonal tumours | Medulloblastomas, molecularly defined Medulloblastoma, WNT-activated Medulloblastoma, SHH-activated and TP53-wildtype Medulloblastoma, SHH-activated and TP53-mutant Medulloblastoma, non-WNT/non-SHH |
| Other CNS embryonal tumours | Atypical teratoid/rhabdoid tumour Cribriform neuroepithelial tumour Embryonal tumour with multilayered rosettes CNS neuroblastoma, FOXR2-activated CNS tumour with BCOR internal tandem duplication CNS embryonal tumour NEC/NOS |
| Pineal tumours | Pineoblastoma |
| Melanocytic CNS tumours | Diffuse meningeal melanocytic neoplasms: melanocytosis and melanomatosis |
| Tumours of the sellar region | Pituitary endocrine tumours Pituitary adenoma/pituitary neuroendocrine tumour Pituitary blastoma |
| Craniopharyngiomas | Adamantinomatous craniopharyngioma |
Neuropathology was one of the first specialties to adopt the integrated diagnostic approach (as part of the ISN-Haarlem Guidelines) when reporting tumour cases.7 Each case is assigned a histological diagnosis, a CNS WHO grade, and a summary of the molecular characteristics, which will be brought together as a final integrated diagnosis (Figure 1).
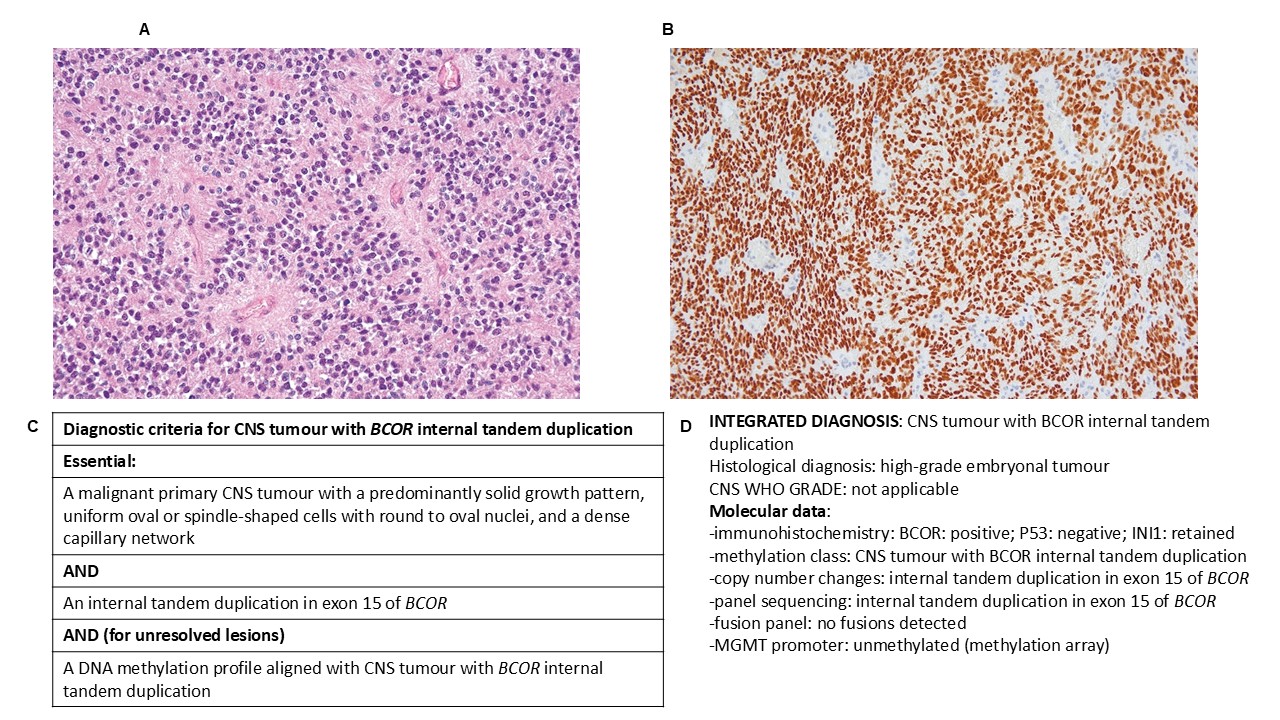
Figure 1. CNS tumour with BCOR internal tandem duplication, a newly defined entity within the WHO classification, frequently seen in children. A. A representative histology image of the tumour. B. Immunohistochemistry demonstrating strong labelling for BCOR. C. The ‘diagnostic criteria’ for this tumour as shown in the WHO classification. Each tumour is provided with a set of essential and desirable features (including molecular) which help to guide diagnosis (5). D. A fictionalised example of the integrated diagnosis for this tumour type, the components of which are provided for each paediatric CNS tumour.
In well-resourced countries, access to such diagnostic tests should be straightforward, e.g. through the Genomic Medicine Service; as an example, for all paediatric brain tumours in England, DNA methylation profiling, DNA NGS sequencing and RNA fusion panel sequencing are funded as standard-of-care. This is not the case internationally, where significant inequalities in access exist. This inequality cascades down treatment pathways. Every child diagnosed with a brain tumour should be able to access such tests irrespective of location. The Asian Oceanian Society of Neuropathology guidelines for Adapting Diagnostic Approaches for Practical Taxonomy in Resource-Restrained Regions (AOSNP-ADAPTR) are designed to help support diagnostics in lower-middle-income countries.8
In well-resourced countries, the issue becomes one of specialisation. Who should report brain tumours? In the UK, most neuropathologists can demonstrate expertise in brain tumour diagnosis, but what is the role of specialist centres and how can they be defined? With only 400–500 cases a year across 43 tumour types and 17 centres, each with 2 or 3 neuropathologists, most neuropathologists will see few, let alone the rarer types and subtypes. The optimal balance of specialist pathology across hospitals, regions and nationally has yet to be resolved.
‘How long will we have to wait?’ is a frequent question in multidisciplinary team meetings. For many brain tumours, the guidelines recommend that adjuvant therapy starts within 28 days of surgery; there is outcome data to support this, in some cases. As turnaround times for molecular tests are significant, often measured in weeks, it becomes challenging to provide an integrated diagnosis in sufficient time for the oncologist to plan and initiate treatment. Furthermore, some patients will require germline testing within this timescale, depending on the pathological classification.
When a diagnosis hinges on the identification of a particular molecular feature, the paediatric neuro-oncology teams and families are in limbo while waiting for molecular testing, unsure how best to proceed without the final diagnosis, exacerbated by the fact that it is not uncommon for molecular tests to change the diagnosis/grade of a tumour. There is no disagreement that molecular tests remain crucial, but refinement of techniques to improve turnaround times is a priority.
Nanopore sequencing may mitigate many of the current challenges around turnaround times. Results can be determined in real-time, with the flexibility to add genes onto a panel while a case is sequenced.9 Currently, intraoperative smear assessment is frequently used by paediatric neurosurgeons seeking an indication of the tumour type, guiding subsequent resection strategies; we are now on the verge of being able to provide a molecular test intraoperatively in parallel with the intraoperative smear assessment, which will be hugely valuable for neuro-oncology teams.
Training existing consultants and the next generation in the latest advances has been important, with an increased appreciation of the need to update training and keep pace. Morphological skills remain vital to training; recognising the tumour type will help guide targeted molecular investigations. However, it is easy to adopt a perception that morphology is less important, as molecular results will provide the diagnosis, leading to less time spent considering the tumour phenotype. Morphology is crucial when deciding and interpreting the molecular results – molecular outputs may not always be correct!
Training the next generation to integrate such results is a new avenue of the training pathway; not all centres have access to molecular testing and, therefore, cases are sent away for analysis, meaning accessing this training can be difficult. For those at the forefront of the integrated diagnosis, gathering feedback about subsequent treatment decisions and outcomes (e.g. did the tumour behave/respond as predicted?) is increasingly difficult. Feedback is an important principle for all work undertaken. It is hoped that with the often-discussed government commitments to improving and integrating national healthcare IT services, this will be better orchestrated. Regular feedback from molecular pathology improves morphological diagnosis and training.
Paediatric brain tumours remain a significant clinical challenge to treat; relapses are common, particularly with higher-grade tumours, and are often untreatable. Most studies have focused on disease at presentation, meaning there is a relative dearth of data about the biology of late-stage and relapsed disease. However, the importance of continual molecular monitoring is now recognised; in the UK, the Stratified Medicine for Paediatrics (StratMedPaeds) trial recruits children, adolescents and young adults with relapsed or refractory disease for multi-modality molecular testing to identify actionable alterations.
There is also an increasing interest among patient and parent groups in exploring brain donation if their child passes away. Several studies have found that families appreciate the opportunity to discuss and consent to an autopsy. Clinicians often avoid discussion of this topic; however, this misses the chance to offer something valued by grieving families. Furthermore, we are missing the opportunity to better understand the molecular characteristics of a tumour in terms of its evolutionary and treatment timeline. This would allow us to appreciate the impact of particular treatments on tumour biology and potentially explore the threat of resistance mechanisms. This can bring some consolation to devastated families after such an awful outcome.10,11
We also need to be mindful of the effect of treatments given for other childhood cancers. Several subtypes of high-grade glioma can occur in the adolescent and young adult years, on a background of treatments for childhood cancers, such as acute lymphoblastic leukaemia or medulloblastomas. How these patients are monitored post-treatment is an important consideration, but also highlights the growing need for safer treatments that do not leave a patient with significant disability, or with the threat of developing a second aggressive tumour.
Paediatric neuropathology is an academic specialty; it is very clear from the significant changes seen across the breadth of the field that research is an integral, not optional aspect of the work. Members of the workforce are either actively engaged in research themselves or provide significant support to other research teams across the UK and internationally via established research networks. Progress is not achievable without a collaborative ethos. Paediatric neuropathology helps to highlight what can be achieved when we work together.
Many neuropathologists are at the forefront of the research advances seen in nanopore sequencing, spatial transcriptomics and tumour classification, among others, and there is a wealth of opportunities to shape an academic career for trainees. However, paediatric neuropathology is not immune to the challenges experienced more generally in academia, including increased financial and time pressures. Therefore, it must be supported as a priority and cornerstone of workforce planning. Neuropathologists act as the bridge between research and clinical teams. It is essential that academia is encouraged as part of a career in this specialty.
Neuropathology is one of the most fast-paced and cutting-edge specialties in pathology and is an attractive area in which to work. There will be many advances in the coming years, which will change practice in paediatric brain tumour diagnostics and shape change in other areas of cellular pathology. Advances in artificial intelligence on the back of a strong foundation in digital pathology are likely to be a key feature. Some in the community fear the impact of this on job security.
However, as with the implementation of molecular advances, neuropathologists will be working with bioinformaticians and clinical, computer and research scientists to guide, advise and even lead the development and implementation of tools that we need on the frontlines of paediatric brain tumour diagnostics, which will also be synergistic with adult brain tumour diagnostic workflows. Consequently, the continual drive to strengthen the link between paediatric brain tumour research and neuropathologists will be a key priority, supporting the transition of data both ways to help the advancement of our vital subspecialty and to lower the significant mortality and morbidity that this disease still has for children.
References available on our website.
Meet the authors
-

Matt Clarke
NIHR Clinical Lecturer in Diagnostic Neuropathology (ST5) at Institute of Cancer Research / University College London Hospitals
Return to January 2025 Bulletin
Read next

Wales Pathology Symposium and coastal walk
20 January 2025

