- Published:
- 15 July 2024
- Author:
- Dr Hilary Longhurst
- Read time:
- 6 Mins
One of the joys of immunology is the opportunity to work with rare diseases and to introduce new and improved treatments. This article explores the extraordinary innovation of in vivo CRISPR-Cas9 gene editing.
Summary
- HAE-C1INH is a rare, disabling condition caused by insufficient regulation of the contact pathway.
- Kallikrein inhibitors are effective in the prophylaxis of HAE attacks.
- NTLA-2002, a CRISPR-Cas 9 targeted at the liver prekallikrein gene, can be delivered in a single, well-tolerated intravenous infusion.
- NTLA-2002 results in ongoing suppression of plasma kallikrein and freedom from HAE-related angioedema.
- Similar CRISPR-Cas9 products could be developed to target other genes and/or organs, providing a new class of drug.
Hereditary angioedema (HAE) is a rare inherited disorder with an estimated prevalence of 1:50,000. It causes episodic angioedema, which can affect the airways (and can result in mortality), gastrointestinal tract (mimicking an acute abdomen and sometimes resulting in inappropriate surgery) and limbs (which can result in disability and inhibited daily functioning).
HAE due to C1 inhibitor (C1-INH) deficiency (HAE-C1INH) occurs because of a heterozygous variant in the SERPING1 gene. The C1 inhibitor regulates several inflammatory pathways, most notably the classical and lectin complement and the fibrinolytic pathways. However, research and clinical experience have demonstrated that lack of C1INH regulation of factor XII and plasma kallikrein of the contact pathway is responsible for the angioedema of HAE-C1INH.
Thus, insufficient C1 inhibitor control of the contact pathway results in massive bradykinin-induced angioedema episodes, which follow minor, usually unknown, local triggers (Figure 1).
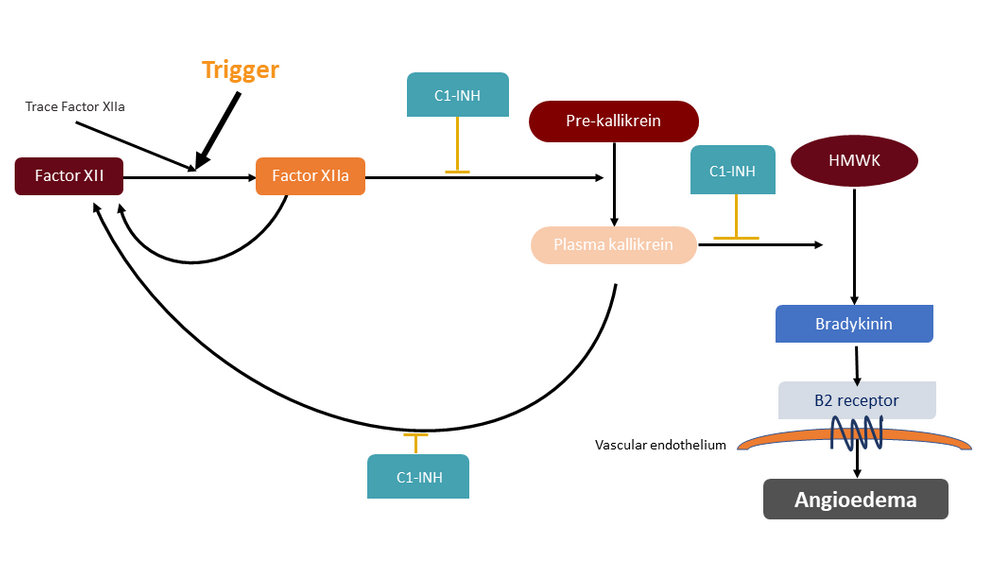
Figure 1: Local low-level factor XII activation occurs constitutively, but if additional activation occurs (for example, by endothelial microdamage) prekallikrein is cleaved to kallikrein, its active form, in sufficent quantities to activate factor XII in a positive feedback loop. Active kallikrein also cleaves high molecular weight kininogen to release bradykinin, which increases local vascular permeability and, if in excess, promotes angioedema.
C1-INH: C1 inhibitor; cHMWK: Cleaved high molecular weight kininogen; HAE: Hereditary angioedema; HMWK: High-molecular-weight kininogen.
Impact of the disease
Many families with HAE have a legacy of premature death; all experience reversible disability, disfigurement and economic and psychological disadvantage. These consequences have largely been hidden from the medical profession. Most angioedemas occur at home and remain undocumented, owing to the ineffectiveness of conventional angioedema treatments, such as antihistamines and corticosteroids, together with the reversible nature of the majority of angioedema attacks.
This is changing. In the last 30 years, the HAE community has organised, forging strong links with their medical advisors and the pharmaceutical industry. This organised and proactive patient group – together with a genetic condition with a known biochemical mechanism and a political desire to facilitate development of drugs for rare diseases – has provided the opportunity for biotech companies to translate new scientific techniques into actual medical treatments for patients with rare conditions, such as HAE. Patients with rare diseases provide an ideal proof of concept for further development for wider indications, while themselves benefiting from the new technologies.
A variety of techniques have been developed to treat HAE-C1INH. Small molecules, monoclonal antibodies and RNA silencing agents that target (pre)kallikrein have demonstrated high levels of efficacy in HAE-C1INH prophylaxis. However, these treatments require lifelong, often parenteral, treatment.
HAE patients in Cambridge, Amsterdam and New Zealand were given the opportunity to participate in a first-in-human trial of an in vivo CRISPR-Cas9 treatment designed to knock down the prekallikrein gene. We predict that this single dose treatment will provide a lifelong cure of HAE symptoms.
The candidate treatment, NTLA-2002, contains a CRISPR-Cas9 RNA encapsulated in a lipid nanoparticle and is administered intravenously. The lipid coating ensures targeted delivery to the liver via its low-density lipoprotein (LDL) receptors, which deliver its CRISPR-Cas9 cargo to the hepatocytes. The CRISPR-Cas9 binds and cuts the hepatic prekallikrein (PKK) gene and reduces systemic kallikrein levels.
As clinicians, we are unused to first-in-human trials, particularly where untested technology is involved. Since HAE is not universally fatal and other treatments exist – though first-line, highly effective treatments are inaccessible to the majority – the threshold for safety is extremely high.
During the development of this treatment, our main concerns were off-site editing, infusion reactions, the long-term lack of plasma kallikrein and the potential of having one’s genes permanently edited with no or only short-acting benefit.
Off-site editing
Off-site editing was the subject of extensive preclinical research.
The target sites of NTLA-2002 were predicted using 2 complementary methods:
- in silico (computational) analysis of the human genome
- in vitro analysis of human genomic DNA.
The potential cleavage sites identified by either of these methods were validated by multiplex polymerase chain reaction (PCR) and next-generation sequencing. The structural variants were assessed by long-range PCR and by short-range PCR with structural variant analysis.
Using these methods, we determined that NTLA-2002 has a sufficiently high level of safety in off-target gene editing.
Additional reassurance was provided from the specific targeting of NTLA-2002 to the liver. Although the liver is the main site of prekallikrein synthesis, other tissues may produce small amounts. The extreme prolongation of activated partial thromboplastin time seen in complete prekallikrein deficiency did not occur in our subjects. Extensively bred mice treated with NTLA-2002 demonstrated no evidence of heritable gene editing.
Long-term lack of plasma kallikrein
Rare individuals with Fletcher syndrome (a congenital complete prekallikrein deficiency) appear to have normal and healthy lives, without excessive thrombosis or bleeding, which provided reassurance.
Lanadelumab and berotralstat, both of which control HAE by suppressing kallikrein levels, have been used in practice for several years, without unexpected safety signals.
Infusion reactions
Preparations were in place for infusion reactions up to and including cytokine storm. We decided on a short premedication regimen of high-dose corticosteroid and – since both the emotional stress associated with treatment and the physical effect of NTLA-2002 could potentially activate the complement pathway – a prophylactic dose of C1INH.
In the end, infusion reactions were short-lived, reversible and relatively mild. We reported reactions including fatigue, low-grade transient fever (on 1 occasion with fleeting shivers) and back pain.
Plasma kallikrein reductions of 60% correlate with almost complete control of HAE attacks in other studies. The results for our first 10 subjects indicated a dose-dependent reduction in plasma kallikrein levels, with sustained mean reductions of 67%, 84%, and 95% for the 25 mg, 50 mg and 75 mg doses, respectively.
Resolution of HAE attacks has been almost complete. After medians of 13.1, 5.7 and 9.3 months follow-up in the 25 mg, 50 mg and 75 mg groups, patients had a reduction and eventual resolution of attacks. They were able to stop prophylaxis without the attacks returning. A patient in the 25 mg arm reported a mild hand angioedema that occurred after a sporting injury at 343 days post-treatment.
The treatment benefit is likely to be long-term. Primate studies have reported continued suppression of plasma kallikrein over 2 years.1,2 Mouse hepatectomy experiments have demonstrated the ongoing presence of edited cells in regenerating daughter liver cells, with continued low kallikrein levels, which suggests that single-dose NTLA-2002 treatment will provide lifelong symptomatic control.
Our subjects are now enrolling in a long-term follow-up study; placebo-controlled trials of the 25 mg and 50 mg doses have begun.The early results suggest that the future is bright for our HAE patients, although long-term follow-up is necessary.
However, in vivo CRISPR-Cas9 technology has far wider potential applications. Other conditions where knockout of a gene in the liver is desirable can be treated by modifying the CRISPR guide. This could potentially deliver a well-tolerated, single-dose treatment.
By modifying the delivery system, other organs could also be targeted. Trials are already in place for transthyretin amyloidosis and a similar in-vivo gene-editing approach could be developed for common conditions, such as some hyperlipidaemias.3
Further reading and references available on our website.
Meet the author
-

Dr Hilary Longhurst
Senior Medical Officer in Immunology, Department of Medicine, University of Auckland and Auckland City Hospital, Te Whatu Ora, New Zealand
Return to the July 2024 Bulletin